Research
We are a computational group — we do not have a wet lab. Every project begins with code and data: genome sequences, protein structures and data. The purpose is to better understand how bacteria and phages co-evolve, and to make predictions that can be verified in the lab. Our experimental validation is done in close collaboration with partner labs.
Our research sits at the intersection of evolutionary biology, microbial genomics and structural bioinformatics. We write Python and R every day. We do not pipette.
The central question
How does evolutionary innovation work at the molecular scale in bacteriophages?
Specifically: how do horizontal gene transfer and recombination generate new receptor-binding protein (RBP) functions, and can we predict — and ultimately design — those functions?
“We are data scientists at heart — but in the scientific sense: we integrate multi-scale data (DNA, proteins, structures, metadata, populations, experimental results) to infer evolutionary processes and find novel patterns in complexity. We have intellectual ownership of our questions. We make direct predictions for experimental collaborators and interpret their results through an evolutionary lens.”
— Rafal Mostowy
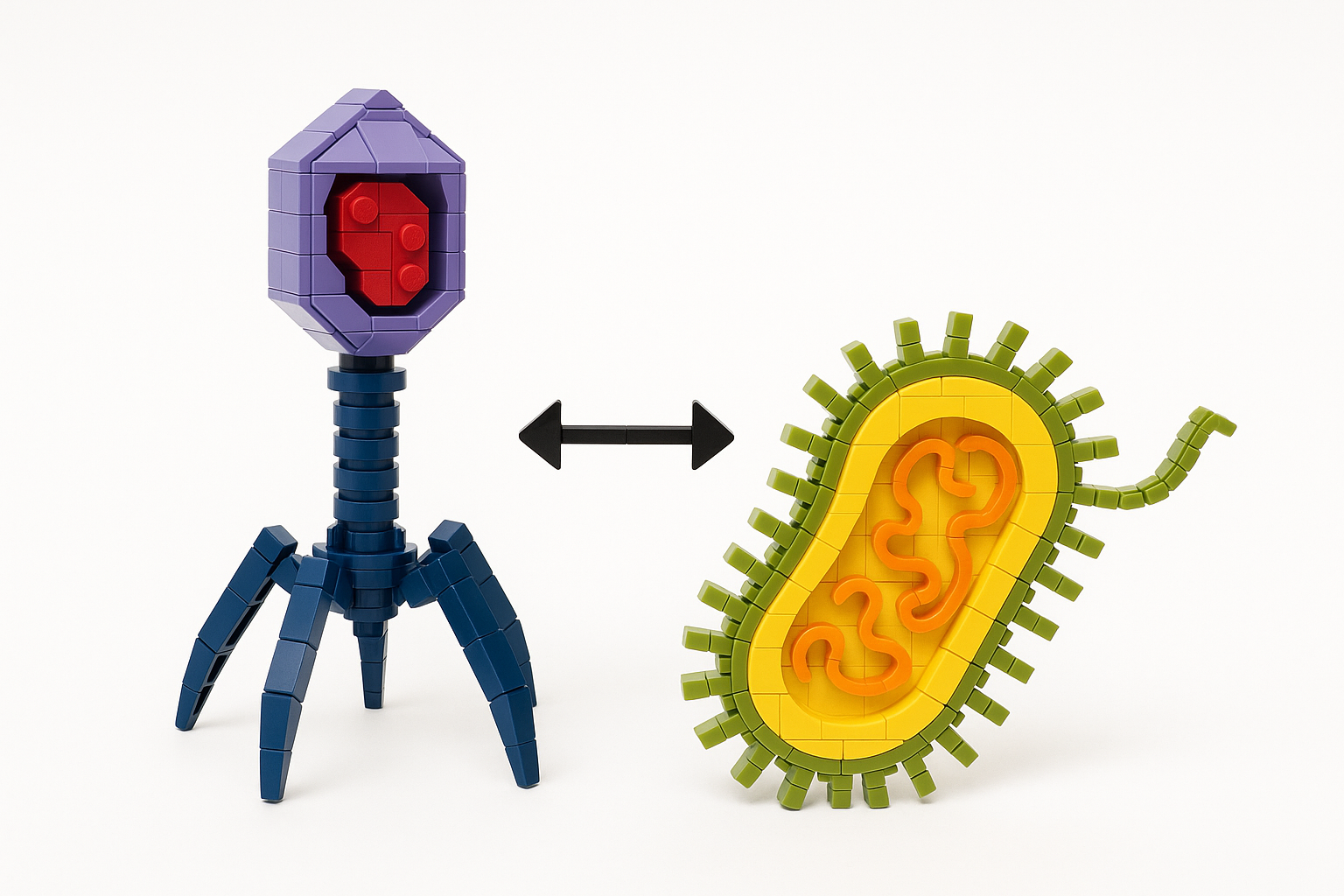
Model system: Klebsiella pneumoniae and its phages
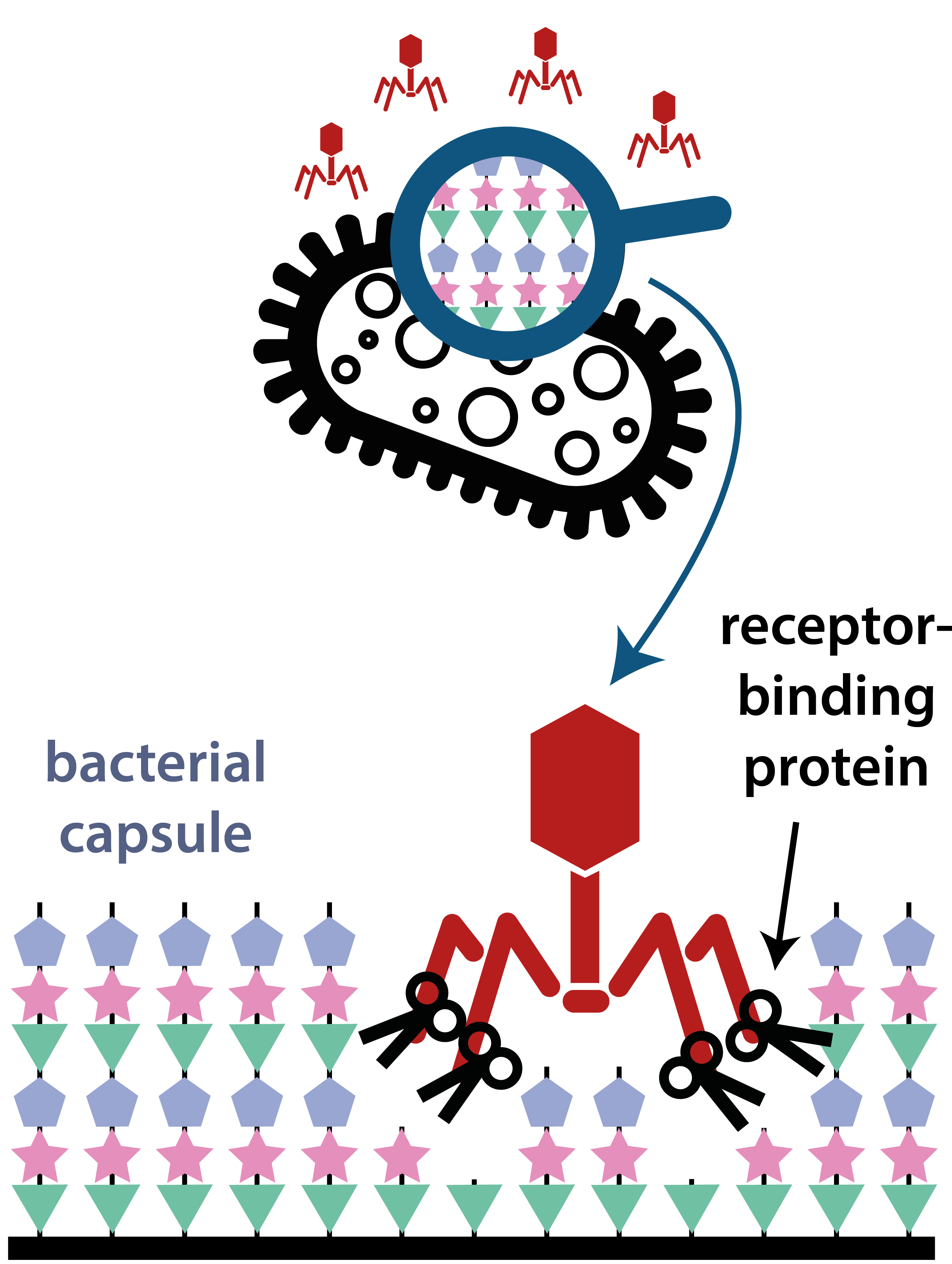
Klebsiella pneumoniae carries a thick polysaccharide capsule — a key virulence factor and antibiotic-resistance enabler. Over 150 distinct capsular (K) types exist, each with a different surface chemistry.
Bacteriophages that infect K. pneumoniae deploy receptor-binding proteins (RBPs) equipped with depolymerase activity. These enzymes are highly specific: each RBP is tuned to recognise and degrade one (or a few) capsule types. That specificity is measurable, the genetic diversity is rich, and the clinical stakes — multidrug-resistant infections, phage therapy — are real.
This makes the K. pneumoniae phage system an ideal platform for studying evolutionary innovation computationally: specificity is measurable, recombination events are traceable, and our predictions can be tested by collaborators in the wet lab.
Three knowledge gaps we are working on
What RBPs exist, and how diverse are they? We are building the first comprehensive catalogue of depolymerase-containing RBPs across Klebsiella phages — combining large-scale genome analysis with structural modelling (AlphaFold3) to map the genetic and structural landscape.
How does sequence and structure map to function? The relationship is more complex than assumed: many RBPs from temperate phages are inactive or have unexpectedly broad specificity. We use comparative genomics and machine learning to link genotype to phenotype, with experimental validation from our collaborators.
How does recombination at the domain and sub-domain level generate new functions? We develop new computational frameworks to detect mosaicism and recombination below the classical domain level, and link these events to functional changes in RBPs.
Key published findings
Protein domain shuffling drives RBP diversity
We analysed over 130,000 representative phage proteins and showed that domain mosaicism — the sharing of functional modules between otherwise unrelated proteins — is a major driver of diversity, with RBPs as a primary hotspot.
Smug et al., Nature Communications 2023. doi:10.1038/s41467-023-43236-9
An ANI gap in bacterial dsDNA viruses
We developed MANIAC, a computational pipeline for accurate average nucleotide identity (ANI) estimation in mosaic viral genomes. Applied to large phage databases, it revealed a multimodal ANI distribution with a gap near 80% — analogous to the bacterial ANI gap, but shaped by viral-specific recombination dynamics.
Ndovie et al., mSystems 2025. doi:10.1128/msystems.01661-24
Capsular specificity in temperate Klebsiella phages
Using a GWAS approach on prophage genomes, we identified diverse structural domains — pectin lyases and SGNH hydrolases — as the determinants of capsule-type specificity. Computational predictions were experimentally confirmed in collaboration with the Drulis-Kawa lab.
Otwinowska*, Koszucki* et al., PLOS Biology 2025. doi:10.1101/2025.06.25.661490
Longer-term vision
Our 10-year goal is a predictive, mechanistic framework for RBP design — grounded in evolutionary principles, not opaque algorithms.
Short term (0–3 years): Catalogue depolymerases for 30–50 K. pneumoniae K-types with experimental validation; establish preliminary design principles at the sub-domain level.
Mid term (3–6 years): Test the hypothesis that bacteria–phage interactions involve a broader enzymatic repertoire (acetyltransferases, SGNH hydrolases beyond canonical depolymerases); refine predictive models to link evolutionary events to function.
Long term (6–10 years): Rational design and proof-of-concept testing of engineered RBPs against clinical K. pneumoniae strains. Translational endpoint: phage therapy and synthetic biology.
Foundational work: bacterial surface diversity
Our earlier work on bacterial polysaccharide evolution directly motivates the phage programme. Surface diversity in bacteria is the evolutionary pressure that drives phage innovation. Key papers:
- Holt & Mostowy (2020). Diversity and evolution of surface polysaccharide synthesis loci in Enterobacteriales. ISME Journal →
- Mostowy & Holt (2018). Diversity-generating machines: genetics of bacterial sugar-coating. Trends in Microbiology →
- Mostowy et al. (2017). Pneumococcal capsule synthesis locus cps as evolutionary hotspot. Molecular Biology and Evolution →
Collaborations
National
- Prof. Zuzanna Drulis-Kawa, University of Wrocław — experimental microbiology, validation of computational predictions
- Dr Stanisław Dunin-Horkawicz, University of Warsaw — computational structural biology
International
- Prof. Kathryn Holt, London School of Hygiene and Tropical Medicine — microbial systems genomics
- Prof. Eduardo Rocha, Institut Pasteur — evolutionary microbial genomics
- Prof. Ed Feil, University of Bath — evolution and ecology of microbial pathogens